|
|||||||||
|
|
Exploring the Genetic Basis of Huntington's Disease
This website was produced as a project for Genetics 677 at the University of Wisconsin-Madison during the Spring 2009 semester. goal of the website is to produce the information I gain from bioinformatics programs in my research the genetic basis of Huntington's disease. If you would like to comment on the site or make a suggestion, please visit the comments page. I am continually looking for information to improve the site.  Courtesy of the Gladstone Institute of Neurological Disease. Background on Huntington's Disease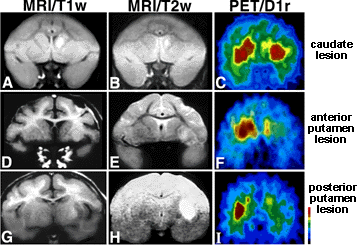
Huntington's Disease (HD) is an autosomal dominantly inherited disorder characterized by progressive onset of neuronal degradation and destruction occurring in certain distinguished areas of the brain. The figure to the right shows a PET brain scan locating lesions in specific locations of a Huntington's disease patient's brain (Image from Harvard Medical School). Primary symptoms of. Symptoms of Huntington's Disease usually become evident from ages 35-45, although it is possible for the disease to manifest itself at any age, including juveniles (Walker, 2007). The earliest apparent symptom of HD is a dramatic shift in a persons mood, including irritability and depression, as reported by the National Institute of Neurological Disorders and Strokes. As the disease progresses, the patient may lose basic short term memory function and the ability to do normal tasks, such as driving. Primary symptoms of Huntington's Disease are chorea, rigidity, dementia, and seizures. Chorea, a characteristic stumbling due to lack of coordination and muscle control, is a main indicator of HD. As neural degeneration progresses, basic functions, such as swallowing or speaking may become impaired. The disease typically lasts 10 to 30 years. Common causes of death are pneumonia, fall related injuries, or other forms of complications. Frequency of the HD is 5-7 per 100,000 individuals in Caucasian populations,, but drops dramatically in Asian populations, to as low as 0.5 cases per 100,000 individuals (Walker, 2007). HD is thus more prevalent in Western countries. The focus of this project is to determine the genetic basis of the Htt mutation and its effects on protein structure and protein function in biological processses. The Huntingtin Gene
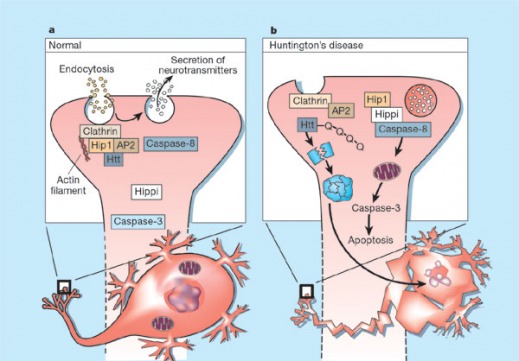
Figure 1: Diagram proposed by Mattson, 2002 of the method in which a polyadenylated huntingtin protein induces apoptosis of neural cells. Diagram A shows normal huntingtin (Htt) binding Hip1, modulating endocytosis. Diagram B shows mutated Htt, which is cleaved by a complex formed by Hip1 and Hippi, forming an aggregate which eventually leads to apoptosis. Huntingtin's Role in Disease
The Htt gene locus is found on the short arm of chromosome 4, specifically 4p16.3, and codes for the Huntingtin protein. The underlying mutation responsible for the disease involves an improper expansion of a CAG trinucleotide repeat region in the gene. While the normal Htt contains 10 to 37 CAG repeats, a patient with a mutated Htt will present anywhere from 38-100 repeat units, resulting in an extended polyadenylate tail on the protein (Gellera, et al. 1996). The quantity of repeat units affects age of onset: a larger number of CAG repeats leads to early manifestation of the disease. People with 36-39 repeat units carry the disease, but most likely will not show symptoms of the HD until around 80 years of age (Rubenzstein, et al. 1996). Normal huntingtin protein is thought to stabilize neurons, preventing apoptosis from occurring and prolonging cell life (Rigamonte, et al. 2000). The mutated huntintin protein is understood to induce apoptosis in neurons, likely as the result of abnormal endocytosis and secretion resulting in a metabolic change in the cell detected by mitochondria, which induces cell death (Figure 1). Currently there is no consensus on the mechanism, although it appears the association of mutant huntingtin with Hip1 and Hippi induces apoptosis (Mattson, 2002). References:  Created by Eric Nickels [email protected] 8/9/2009 Genetics 677 Webpage |
||||||||
|
|
|||||||||
