|
|||||||||
|
|
This website was created as a project for Genetics 677 at UW-Madison in the spring of 2009 Trinucleotide disorders
Huntington's disease is one of many disorders caused by a trinucleotide repeat mutation. Below I will present some of the other known trinucleotide disorders and their genetic basis in order to demonstrate the similarities and differences between huntingtin and the variety of other diseases. Fragile-X Syndrome
Fragile-X syndrome results from a trinucleotide repeat expansion in an X-linked gene, FMR1, or fragile-x mental retardation 1 protein (also described as FMRP). FMR1 is characterized as an RNA-binding protein which is important in RNA localization within the cell (data from UniProtKB). A CGG repeat section immediately preceeding the coding region acounts for the expanded region capable of inducing the disease (OMIM). The disease results from an expansion of 200 or more CGG repeats. Interestingly, an expansion of 50-200 repeats leads to a different disorder, fragile X tremor/ataxia syndrome. This still results from disruption of the FMR1 gene, with the repeat section termed a "premutation (OMIM). The disorder results from the abnormal methylation of this preceeding region, which actually blocks transcription of the FMR1 gene (OMIM). Thus, the disease results from a loss of function in the FMR1 locus, which is quite departed from the gain of function proposed for the intranuclear inclusions created by the cleavage of mutant huntingtin in Huntington's disease. An interesting approach to take with this disease, in my opinion, would be to analyze the DNA methylating proteins which account for the blocked transcription of FMR1. 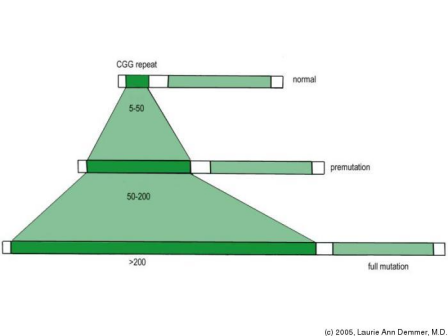 Above: Depiction of the role of CGG repeat sections in development of fragile X syndrome (full mutation), fragile X tremor/ataxia (premutation), and a wild type gene. Image retrieved from http://ocw.tufts.edu/Content/20/lecturenotes/301035/301049 Dentatorubral-pallidoluysian atrophyDRPLA is caused by a similar trinuclear repeat mutation to Huntington's disease in that it is a CAG expansion. The protein affected in DTPLA is atrophin-1, which is uncharacterized in its function although is beleived to be involved intranscriptional co-repression. This disease is of particular interest to this project, as it results from a similarly expanded CAG region and the proteolytic cleavage of the subsequent expanded protein. According to OMIM, symptoms of the disease include myoclonic epilepsy, dementia, ataxia, and choreoathetosis. The disease was originally discovered in Japan, where the prevalence is highest, although other forms of the disease emerged in other locations, in particular Haw River, NC, and the UK. These other locations were originally described as different diseases, as the phenotypes differed to a certain degree, until genetic research indicated the varying degrees of CAG repeats lead to different phenotypic manifestationsof the disease (OMIM). The CAG repeat region may be expanded from a normal range of 7-23, or to a pathological expansion of 49-75. The cleaved atrophin-1 protein also forms intranuclear inclusions, which bind transcription factorts and lead to dysregulation of a variety of genes. The result is, eventually, atrophy of neuronal tissues in the CNS. SummaryThe study of various trinuclear disorders presents an interesting insight into Huntington's disease and the role of intranuclear inclusions formed by the cleaved mutant protein. As demonstrated by the two disorders above, trinuclear disorders in general do not necessarily induce the same effects upon cells, especially in the case of the more distantly related fragile X syndrome and the CGG repeat. Yet in DRPLA, the same type of expansion leads to a similar method of pathogenesis. Studying other CAG repeat disorders may present some of the mechanisms of Huntington's disease. If a polyglutamine tract of atrophin-1leads to nuclear inclusions which block normal transcription, how do these structures differ from the intranuclear inclusions of huntingtin, which similarly block normal transcription? Studying the structure of these aggregates may indicate the basis of the phenotypic differences in the diseases. Also, the normal functions of atrophin-1 and huntingtin both remain poorly defined. Studying the normal function of each protein may provide even more interesting clues about the disorders which may one day lead to a suitable therapy for recovery of affected patients. Other disorders resulting from CAG repeats exist as well, including the X-linked spinal and bulbar muscular atrophy or Kennedy disease and spinocerebellar ataxia. Created by Eric Nickels [email protected] 5/9/2009 Genetics 677 Webpage |
||||||||
|
|
|||||||||
