|
|||||||||
|
|
This website was produced as an assignment for Genetics 677 at UW-Madison Spring 2009 Phenotypes
I used RNAi databases for C. elegans and D. melanogaster to research phenotypes of human huntingtin homologs.The most similar gene to human huntingtin in C. elegans is the orthologous gene F21G4.6. I used F21G4.6 to conduct a search of RNAi knock out experiments to detrermine the corresponding phenotypes under regulation of the gene. Results from the two programs listing results for F21G4.6, Wormbase and Phenobank , are shown below. Other programs, including RnaiDatabase, did not return results for the gene. For D. melanogaster, I found results at Flybase.org. Wormbase
Wormbase.org provided the results of RNAi knock out experiments for F21G4.6. Although the site lists multiple RNAi experiments analyzing 10 different phenotypes, there are no observable results from the RNAi. The site lists the 10 phenotypes not observed, shown below. Phenotypes not observed 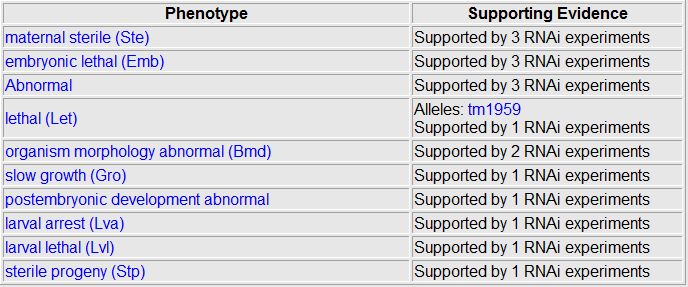 Phenobank
In contrast, the Phenobank database shows 1 known phenotype derived from RNAi experiements. In contrast to the other databases, Phenobank shows sterility F0/fertility problems as a phenotype for F21G4.6. The site notes the phenotype is of low reproducibility, but high penetrance. The dsRNA utilized for the knockout experiments was listed as dsRNA 67-e3, shown below. This leaves me less confident in the results. Of the 3 tests conducted, only one gave the sterility phenotype, while the data on one test was inconclusive and the final test was wild type.  Left: F21G4.6 RNAi results in sterility 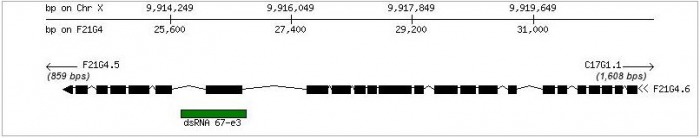 F21G4.6 and dsRNA 67-e3 FlyBase
Flybase.org contained a listing for a gene similar to human huntingtin, CG9995 or Dmel/htt, in D. melanogaster. The site contained a large amount of information regarding this gene, including gene ontology and orthologous genes in other eukaryotic organisms, which demonstrated similarity of the CG9995 gene to those I researched for huntingtin gene homolgy. Concentrating on the phenotypes controled by CG9995, I found alllelic phenotypes listed, shown in the table below. The four phenotypes are listed as axon./larval stage, eye, larval brain, and ommatidium. This information indicates the gene in D. melanogaster is active in neuronal tissue, similar to human huntingtin. The ommatidium is a single unit of the compound eye of insects. Thus, all of the phenotypes listed correspond to neurons or sensory organs. Phenotypes listed for CG9995 on Flybase 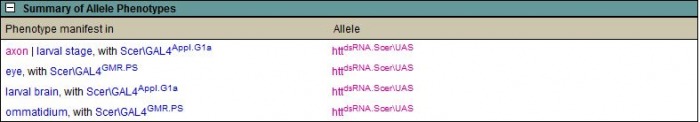 ZfinZfin provides data on genes annotated in Danio rerio. The homologous gene to human huntingtin in Danio reiro, also denoted htt, demonstratelisted the phenotypic effects of RNAi resulting from research conducted in 2007 by Lumsden, et al. The phenotypes listed were observed in blood, brain extension, eye, growth, head, pericardium, pigment accumulation, post-vent region, ventricular system, and the whole organism. Lumsden, et al. observed a disruption in the normal iron homeostasis in specimens exhibiting knocked out htt function (1). The research demonstrated cells were incapable of utilizing endocytosed Fe++ molecules through the normal transferrin mediated pathway, although iron molecules were still present in the cells. This suggested the loss of normal htt function prevents the unloading of Fe molecules from the endocytosed vesicles, and points toward a deficiency in normal cell metabolism, which is required in the oxidative pathway (1). Since iron accumulation and decreased neuronal metabolism are both features of Huntington's disease, the paper suggests there may exist a link between the disrupted iron transfer capability of htt deficient organisms and pathogenesis of the disease (1). A schematic of the disrupted pathway is diagrammed below. Right: Schematic proposed by Lumsden et al. (1) for the role of Danio reiro htt in iron homeostasis. Gene knockout experiments conducted with htt demonstrated a deficiency in the endocytosis pathway of iron. The authors suggest htt plays a role in the unloading of iron from endocytic vesicles. The implications of this deficiency may prove important in the overall maintanence of metabolism in affected cells, as aerobic respiration provided by mitochondria require iron. 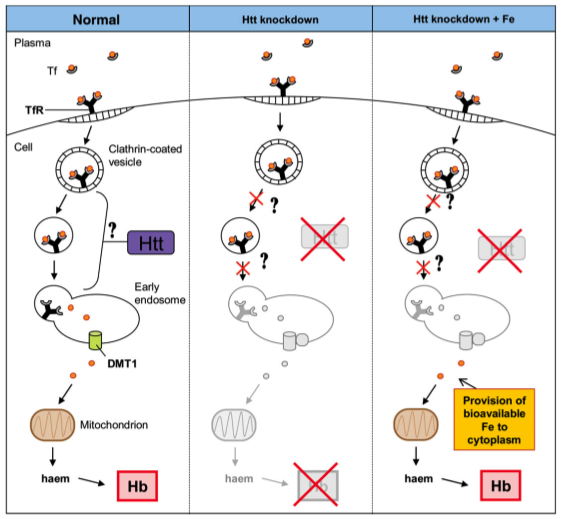 MGI mouse models
MGI, a mouse genome database, lists a total of 27 mouse models for Huntington's disease resulting from different types of genetic manipulations, such as knock in and knock out models. One of the models, MGI:3583991or mhtt-exon1 (103Q) is a knock-in mouse utilizing the loxP system to induce a premature stop codon into the normal mouse htt gene. This leaves the mouse with a cleaved huntingtin protein, which is similar to the human huntingtin disease modification. The phenotypes resulting from this knock in are somewhat similar to human Huntington's disease: Abnormal pyramidal neuron morphology, gliosis, abnormal inhibitory postsynaptic currents, and neurodegradation. Hyperactivity was also noted as a behavioral change. This model, thus, appears to be a good representation of Huntington's disease in mice. Results
The data from the two C. elegans RNAi databases does not provide convincing support for phenotypes derived from the huntingtin ortholog F21G4.6. The overwhelming consensus of no observable phenotypes from Wormbase combined with the relatively low suppor for sterility/fertility problems from Phenobank lead me to believe RNAi in C. elegans may not be useful, currently, concerning this gene. Of course, a phenotype such as sterility is likely difficult to diagnose as a resarcher, and perhaps the results from Phenobank only indicate more experiments must be conducted on the genel. The data from Phenobank is, of course, still relevant, even with a paltry amount of support. I found the flybase results more interesting, as the observed phenotypes suggest a linkage of the gene to human huntingtin, which similarily affects neuronal tissue. (1) Lumsden et al. 2007. Huntingtin deficient zebrafish exhibit defects in iron utilization and development. Human Molecular Genetics Vol. 16, No. 16. 1905-1920. doi:10.1093/hmg/ddm138 Created by Eric Nickels [email protected] 5/9/2009 Genetics 677 Webpage |
||||||||
|
|
|||||||||
